Motivation¶
The aim of this package is to provide software tools for predictive modeling of the long timescale dynamics of biomolecular systems using statistical modeling to analyze physical simulations.
Given a dataset of one or more stochastic trajectories tracking the coordinates of every (10,000+) atom in a molecular system at a discrete time interval, how do we understand the slow dynamical processes and make quantitative predictions about the system?
Workflow¶
To build a dynamical model, we apply (stepwise) a series of dimensionality reductions. The basic set of steps is outlined below. Note that most steps are optional under certain circumstances. The particulars should become clear as you continue reading the documentation.
- Set up a system for molecular dynamics, and run one or more simulations for as long as you can on as many CPUs or GPUs as you have access. There are a lot of great software packages for running MD, e.g OpenMM, Gromacs, Amber, CHARMM, and many others. MSMBuilder is not one of them.
- Featurize trajectories into an appropriate vector of features. The full \(3N\) set of atomic coordinates is potentially unwieldy and redundant. It likely does not respect the rotational or translational symmetry of your system either. We commonly use backbone dihedral angles as our features, although this depends highly on the system being modeled.
- Decompose your features into a new basis that preserves the relevant information in your data with fewer dimensions. We typically use tICA, which finds linear combinations of input degrees of freedom that maximize autocorrelation or “slowness”.
- Cluster your data to define (micro-)states by grouping similar input data points. At this stage, we’ve reduced the dimensionality of the problem from potentially thousands of \(xyz\) coordinates to a single cluster (state) index.
- Estimate a model from the clustered data. We typically build an MSM, which models the important dynamics of the system.
- Use GMRQ cross-validation to select the best model. There are many hyperparameters (knobs to tweak) in the workflow. This scoring function can help us pick the best values.
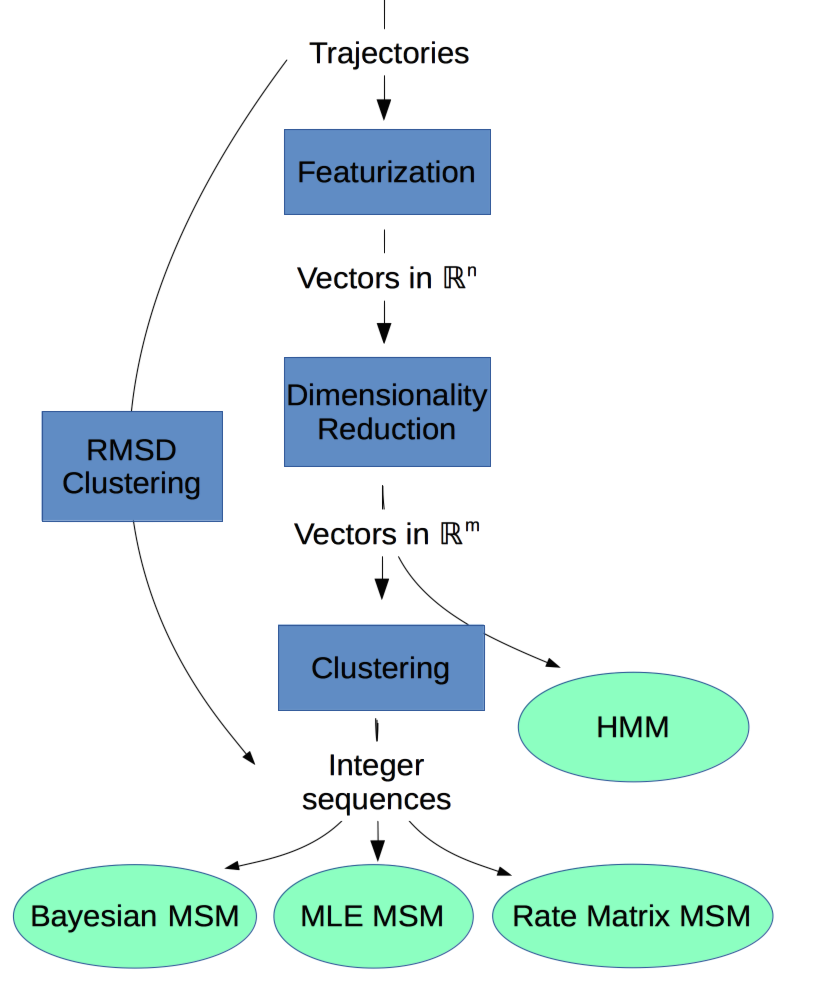
A diagram of potential workflows.